La plateforme « Regards et débats sur la biodiversité » de la Société Française d’Ecologie (SFE) a maintenant trois ans. La SFE vous propose cette semaine le regard n°50 : c’est celui de Thibaud Decaëns, David Porco et Rodolphe Rougerie, chercheurs en écologie et en zoologie, sur la méthode du barcoding ADN et ses multiples applications à l’étude de la biodiversité.
MERCI DE PARTICIPER à ces regards et débats en postant vos commentaires et questions après ces articles; les auteurs vous répondront.
Le barcoding ADN :
un outil pour étudier la biodiversité des invertébrés terrestres
Thibaud Decaëns(1), David Porco(1) et Rodolphe Rougerie(2)
(1) Laboratoire ECODIV, EA 1293, Université de Rouen
(2) Unité de Zoologie Forestière, INRA d’Orléans
———
Mots clés : biodiversité, espèces, taxonomie moléculaire, diversité cryptique, méthodes, barcoding ADN,
barcoding environnemental, invertébrés terrestres.
Introduction : Les invertébrés terrestres, une grande diversité d’espèces à explorer
Les invertébrés terrestres, c’est-à-dire les animaux non vertébrés vivant dans les écosystèmes terrestres, sont de loin les organismes les plus diversifiés de notre planète (Gullan & Cranston 2010). On y dénombre des millions d’espèces d’insectes et d’autres formes de vie remplissant de nombreuses fonctions écologiques et interagissant de façon contrastée avec les sociétés humaines (voir le regard n°28 de S. Barot et F. Dubs sur cette plateforme). Alors que certaines participent à la production de ressources ou de « services environnementaux » bénéfiques aux sociétés humaines (cf. les regards n°4, n°30 et n°46), d’autres se distinguent au contraire par leur capacité à s’attaquer aux cultures ou à véhiculer des pathogènes transmissibles à l’homme (voir par ex. les regards n°18 et n°24).
Malgré leur importance écologique et économique, la connaissance systématique (taxonomique*) de ces organismes est encore très insuffisante. A l’heure actuelle, nous n’avons probablement pas découvert plus de 20% de leur diversité mondiale (Primack 2000), et cet important déficit taxonomique* représente un frein considérable pour toute étude visant à décrire leurs patrons de biodiversité quelle que soit l’échelle considérée (Janzen 2004; Condon et al. 2008; Decaëns 2010; Janzen 2010, et voir le regard n°23).
Si les connaissances sur la taxonomie et la répartition géographique (biogéographie) des invertébrés sont encore embryonnaires, c’est parce que leur identification soulève de nombreuses difficultés. Parmi celles-ci on peut noter (1) une variabilité importante des caractères morphologiques utilisés pour les identifications ; (2) des niveaux élevés de diversité (génétique) non morphologique, dite cryptique ; (3) le caractère incomplet des clés ou guides d’identification, lorsqu’ils existent, du fait du manque de connaissances sur la diversité des invertébrés (tout particulièrement en région tropicale) ; (4) une inadéquation de ces clés d’identification vis-à-vis de certains stades larvaires ou de certaines castes* d’individus, chez les espèces sociales ; (4) l’exigence d’un niveau d’expertise scientifique élevé pour l’utilisation de ces clés ; (5) une raréfaction progressive des taxonomistes spécialistes de ces groupes, au sein de la communauté scientifique (Decaëns 2010).
Ainsi, nous sommes toujours incapables de quantifier avec précision le nombre d’espèces d’invertébrés qui peuplent notre planète, d’identifier les menaces qui pèsent sur ces organismes et de prédire l’évolution de leurs communautés face aux changements globaux actuels (Wilson 2002). A d’autres échelles, il est encore difficile de décrire avec exactitude la diversité et la structure des communautés locales d’invertébrés, notamment dans les régions tropicales dont les faunes sont à la fois les plus riches et les moins bien connues.
Cependant, une technique d’identification biomoléculaire assez récente, le « barcoding ADN », devrait largement dynamiser les recherches en taxonomie et donc favoriser les progrès des connaissances sur la diversité des invertébrés, la dynamique de leurs communautés, leur rôle dans les écosystèmes et leur évolution.
.
La méthode du barcoding ADN
Il y a dix ans, Hebert et al. (2003) proposaient pour la première fois le concept de « DNA barcoding » ou « code-barres ADN ». L’approche consiste à utiliser un fragment standard du génome* comme marqueur génétique pour la discrimination des espèces. Le fragment choisi (du moins pour le règne animal) est un morceau du gène dit CO1, un gène mitochondrial* codant pour une sous-unité d’une enzyme clé dans les chaînes de réactions biochimiques de la respiration (métabolisme aérobie), la Cytochrome c Oxydase.
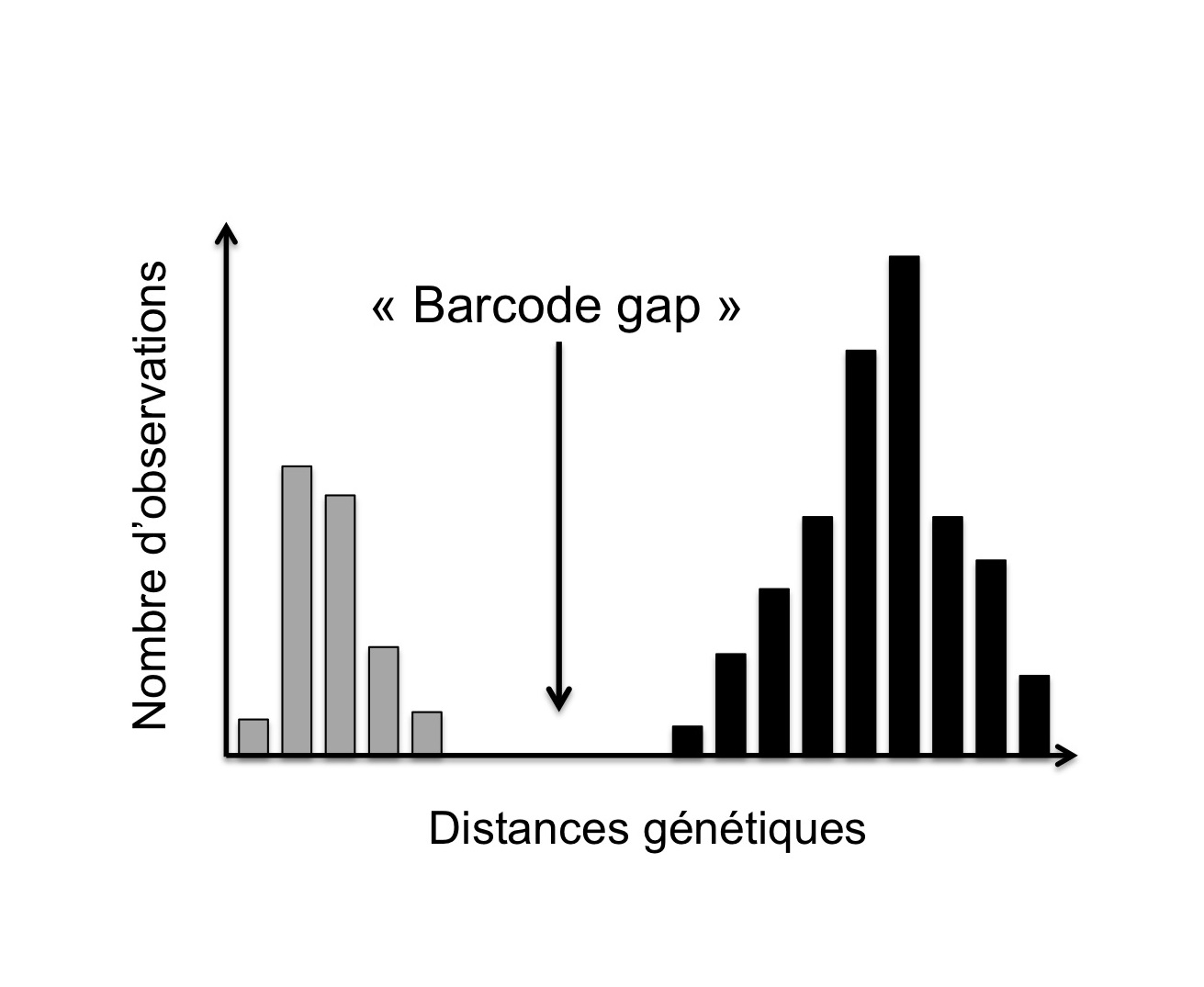
Figure 1. Illustration du « barcode gap »: distribution théorique (diagramme de fréquences) des distances génétiques à l’intérieur des espèces (i.e. intra-spécifiques, barres grises) et entre espèces (i.e. inter-spécifiques, barres noires) au sein d’une population d’individus composée de deux espèces proches (d’après Decaëns et al., 2013).
(1) sa vitesse d’évolution, suffisamment rapide en général pour permettre l’accumulation substantielle de mutations (souvent neutres) et donc la différentiation génétique d’espèces et sous-espèces ;
(2) une différence marquée entre la variabilité observée entre individus d’une même espèce et celle observée entre individus appartenant à des espèces différentes (notion parfois désignée comme le « barcode gap », Figure 1) ;
(3) la relative facilité de sa multiplication (amplification par PCR*) et de son séquençage, du fait de l’existence d’une multitude de copies dans les cellules et de régions relativement conservées du gène permettant le développement et l’utilisation d’amorces dites « universelles », utilisables pour une grande variété d’organismes.
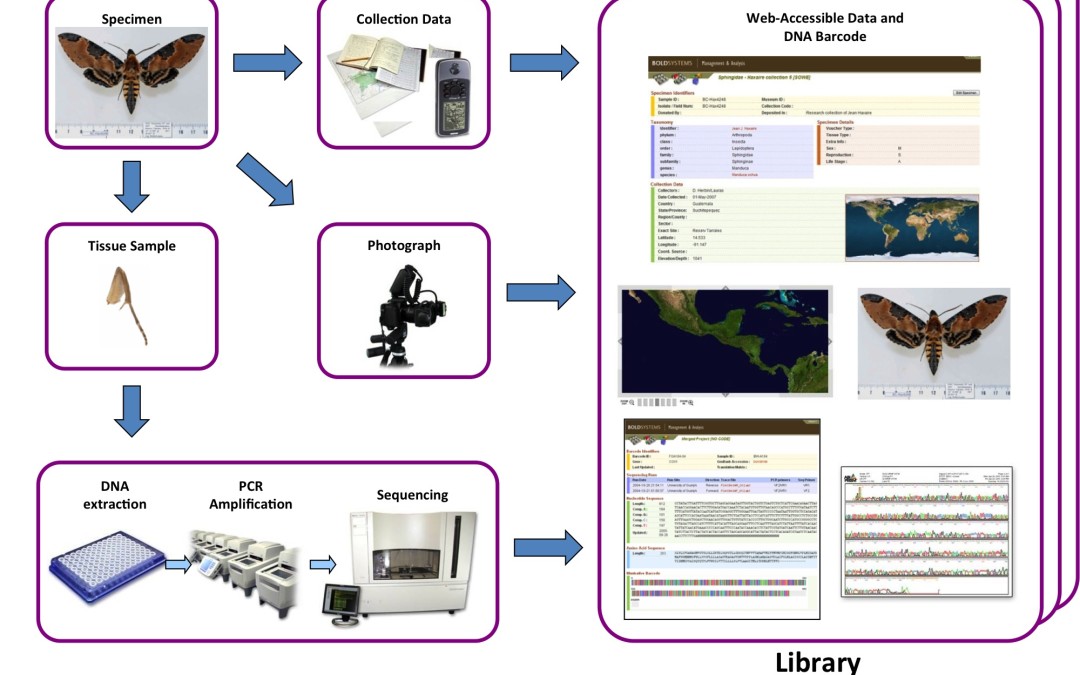
A condition d’avoir accès à une ou plusieurs bibliothèques de références, où les séquences d’ADN sont associées à une identification fiable (basée sur la morphologie et réalisée par un spécialiste), un chercheur ou un enquêteur sans compétence en systématique peut utiliser cette méthode de barcoding pour identifier un organisme d’une espèce inconnue de lui : il lui suffit pour cela de déterminer la séquence de cette région du gène CO1, selon la méthode de Hebert et al., et de la comparer aux séquences présentes dans ces bibliothèques (cf. Figure 2 ci-dessous).
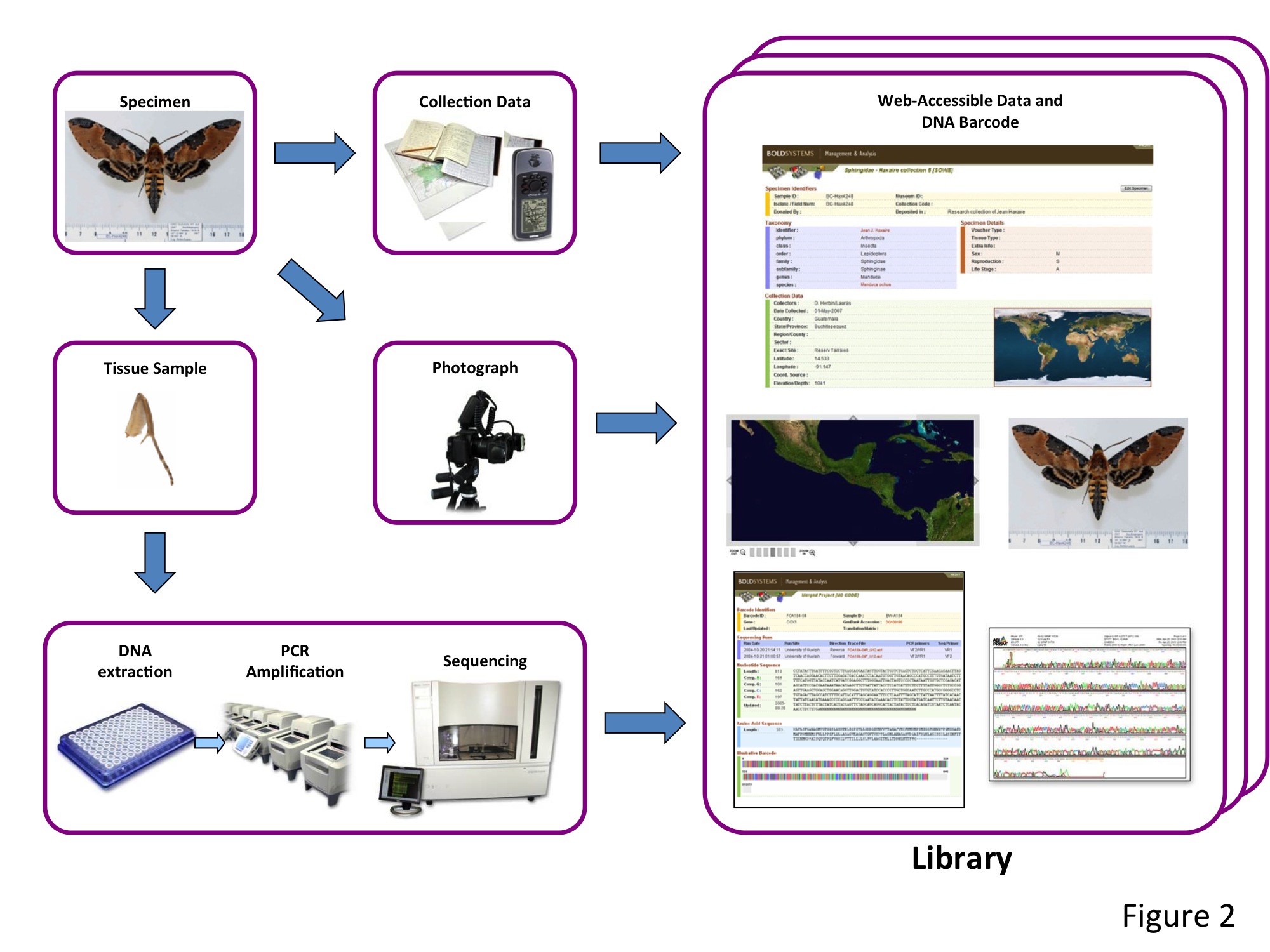
Figure 2. Les différentes étapes nécessaires pour l’obtention du code-barres d’un spécimen et son dépôt dans une librairie de référence. Chaque séquence ainsi obtenue est associée à diverses données dont un nom d’espèce binominal en latin attribué par un expert, une photo et des données relatives à la localité de collecte du spécimen.
Grâce à cette méthode, l’utilisateur peut lire et identifier cette séquence clé de la même façon que l’on scannerait le code-barres d’un produit dans un supermarché pour en connaître l’intitulé ou le prix. Le code-barres ADN mène ainsi l’utilisateur non expert au nom latin associé à cette séquence dans la bibliothèque, ainsi qu’à toutes les autres informations associées qui peuvent s’y trouver (images, distribution, données écologiques, etc.).

Figure 3. Le symbolisme du code-barres ADN s’inspire des codes-barres utilisés dans les supermarchés pour connaître instantanément le prix ou l’intitulé d’un produit. Le code-barres ADN d’une espèce dans une librairie de référence va ainsi mener l’utilisateur au nom latin associé à cette séquence ainsi qu’à l’ensemble des informations qui s’y rattachent
(Figure d’après Stoeckle & Hebert, 2008).
Avantages et limites de la méthode
Les avantages du barcoding ADN sont multiples. Cette méthode :
(1) est reproductible et testable tant qu’est maintenu le lien entre chaque séquence d’ADN et un spécimen de référence ;
(2) permet des identifications massives et rapides (immédiates dès l’obtention des séquences) de spécimens, offrant une alternative aux identifications morphologiques classiques fastidieuses, longues et coûteuses ou parfois même impossible faute de spécialiste disponible ;
(3) est accessible à tous et partout, dès lors que des moyens de séquençage sont disponibles ;
(4) permet de traiter toutes sortes de spécimens, y compris ceux non-identifiables par la morphologie (œufs, larves, fragments de tissu, etc.).
Certaines études ont cependant mis en avant les limites de la méthode et les risques potentiellement associés à son utilisation, parmi lesquels :
(1) l’absence de différences génétiques diagnostiques entre certaines espèces pourtant distinctes au plan morphologique, du fait par exemple d’un temps de divergence trop court ou d’un phénomène d’introgression* entre les espèces ;
(2) de fortes variations génétiques au sein d’autres espèces, pouvant suggérer l’existence d’espèces distinctes, mais résultant plutôt de l’existence d’une forte variabilité génétique entre populations géographiquement isolées ;
(3) le risque d’amplification de pseudogènes* (ou copies nucléaires) qui peuvent engendrer des complications méthodologiques et affecter la fiabilité des résultats.
La meilleure solution pour éviter ces pièges réside dans l’utilisation combinée du barcoding et d’autres jeux de données (morphologie, autre marqueur génétique, données écologiques ou biogéographiques) permettant d’augmenter la robustesse des interprétations. En l’absence de données corroboratives, le barcoding ADN doit être utilisé avec les précautions nécessaires liées à l’utilisation d’un marqueur moléculaire unique.
Encadré : Les campagnes internationales de barcoding
Le barcoding ADN est l’objet de différentes initiatives nationales et internationales, parmi lesquelles la plus ambitieuse est le projet « International Barcode of Life » (iBOL, http://ibol.org/), qui a permis de mobiliser un large réseau international de spécialistes avec comme objectif de construire une librairie de cinq millions de codes-barres ADN pour 500 000 espèces d’ici à 2015. Le projet est structuré en groupes de travail portant sur des régions précises du globe ou sur des groupes taxonomiques ou fonctionnels prioritaires parmi lesquels de nombreux groupes d’invertébrés terrestres. Ainsi les vers de terre, les fourmis ou encore les lépidoptères sont l’objet de campagnes spécifiques dont les résultats sont présentés dans des sites internet dédiés (Figure 4).
Figure 4. Le site web « Formicidae Barcode of Life » présente l’avancée et les principaux résultats de la campagne de barcoding ADN des fourmis du Monde
(http://www.formicidaebol.org/).
L’ensemble des codes-barres générés est compilé dans une plateforme bioinformatique accessible à tous, le « Barcode of Life Data Systems » (BOLD, http://www.barcodinglife.org) (Ratnasingham & Hebert 2007), qui permet aux utilisateurs de comparer les séquences entre elles et de réaliser les premières analyses nécessaires à leur valorisation scientifique. À ce jour, plus de deux millions de séquences sont stockées dans la base de données, représentant environ 170 000 espèces formellement identifiées (voir le site de BOLD pour un chiffrage actualisé). Les taxonomistes jouent un rôle clé dans la construction et la gestion de cette librairie, car ils sont les seuls à posséder l’expertise nécessaire pour valider les résultats du barcoding ADN, identifier les éventuels problèmes et valoriser les résultats au travers de descriptions d’espèces nouvelles ou de révisions taxonomiques. BOLD est également une plateforme de travail en ligne utilisée quotidiennement par des chercheurs dans le monde entier, combinant des données publiques et accessibles à tous, et des données gardées privées durant la période d’analyse et de valorisation par les chercheurs participant aux campagnes. Un outil d’identification est également présent sur BOLD, permettant de comparer la séquence d’ADN d’un organisme inconnu à l’ensemble des codes-barres ADN présents dans la base de données.
Applications à l’étude de la biodiversité des invertébrés terrestres
Diversité cachée, ou cryptique
De nombreuses études récentes utilisant la variabilité de l’ADN pour explorer la diversité des invertébrés ont mis en évidence l’existence d’espèces clairement individualisées d’un point de vue génétique mais pourtant indissociables par leur morphologie. Ces espèces ont été qualifiées dans la littérature d’espèces cryptiques (Hebert et al. 2004).

Figure 5. Exemple de diversité cryptique chez les vers de terre : les deux espèces Lumbricus terrestris (à gauche) et Lumbricus herculeus (à droite) ont longtemps été considérées comme une seule et même espèce jusqu’à ce que l’étude de leurs codes-barres ADN ne prouve le contraire (James et al., 2010). (Cliché : T. Decaëns)
Chez les vers de terre, 30 % en moyenne des espèces définies d’après leur morphologie seraient ainsi des complexes d’espèces cryptiques (Decaëns et al. 2013). D’une façon similaire, Janzen et al. (2013) ont montré que 32 espèces morphologiques supposées bien connues de papillons de nuit (famille des Saturniidae) de l’Aire de Conservation de Guanacaste (Costa Rica) représenteraient en fait 49 espèces véritables.
La multiplication des études utilisant le barcoding ADN fournit un nombre croissant d’arguments permettant de supposer que des niveaux équivalents de diversité cryptique existent chez la majorité des groupes d’invertébrés terrestres (oligochètes, collemboles, lépidoptères, coléoptères, etc.).
Ces résultats suggèrent que les estimations des niveaux globaux de biodiversité, qui se sont traditionnellement appuyés sur des connaissances taxonomiques basés sur la morphologie, devraient être revues à la hausse.
L’existence d’espèces cryptiques remet également en question la validité de certaines connaissances scientifiques qui pourraient être basées sur une classification mal comprise. Certaines espèces considérées à tort comme indiscutables, telles que Lumbricus terrestris, ont été l’objet d’un nombre considérable d’études scientifiques relevant de domaines scientifiques aussi variés que l’écologie, l’éthologie, la physiologie ou la biologie moléculaire. Les résultats et conclusions obtenus par ces études sont donc potentiellement biaisés, car ces espèces cryptiques sont susceptibles de différer de façon substantielle dans un certain nombre de leurs caractéristiques.
Omettre cette diversité cryptique pourrait donc sérieusement entraver les efforts des biologistes et des écologues visant à mettre en œuvre des études comparables et à proposer des généralisations fiables. Il existe un sérieux risque de construire une « biologie chimérique » (Porco et al. 2012a, 2012b; Decaëns et al. 2013), dans laquelle seraient décrites sous un même nom d’espèce erroné les caractéristiques de plusieurs espèces véritables.
Taxonomie
L’utilisation du barcoding ADN dans des actes taxonomiques formels (description de nouvelles espèces, révisions taxonomiques) a augmenté de façon constante au cours des dernières années. L’exemple déjà cité de L. terrestris et L. herculeus illustre parfaitement la façon dont cet outil peut mettre en lumière des séparations entre espèces proches que la morphologie seule n’avait pas été capable de détecter. De nombreux autres exemples de ce type existent, où le barcoding ADN a permis d’identifier des séparations entre espèces cryptiques, permettant ainsi leur description (dans le cas de taxons nouveaux) ou leur réhabilitation (dans le cas d’espèces ayant été décrites par le passé puis mises en synonymie de façon erronée).
Dans d’autre études, les codes-barres ADN sont simplement utilisés en complément d’autres caractères dans la description d’espèces nouvelles aisément séparables par ailleurs (sur la base par exemple de leur morphologie), ou dans des révisions taxonomiques plus larges portant sur des groupes d’espèces ou des genres particuliers. Ils apportent simplement dans ce cas des arguments complémentaires aux scientifiques pour conforter leur décision de créer des nouveaux noms scientifiques ou de redéfinir les contours de la classification taxonomique tels que les avaient définis leurs prédécesseurs.
D’une façon générale, de plus en plus d’auteurs recommandent l’utilisation systématique des codes-barres ADN dans les descriptions d’espèces nouvelles ou les révisions taxonomiques. Une telle généralisation de l’outil permettrait notamment d’associer une séquence à chaque spécimen type pour toute nouvelle espèce décrite, et de rendre disponibles ces informations au travers des librairies de référence pour les études à venir. Par ailleurs, les progrès méthodologiques pour le séquençage de l’ADN ancien permettent désormais d’accéder à des informations génétiques pertinentes pour des spécimens conservés en collection depuis plusieurs siècles. Ces avancées sont particulièrement intéressantes pour l’exploitation des collections muséales (Puillandre et al. 2012) et plus particulièrement pour les spécimens types dont les codes-barres ADN, même partiels, peuvent s’avérer indispensables pour résoudre de manière non équivoque des questions de nomenclature autrement insolubles (James et al. 2010; Rougerie et al. 2012).
Phylogéographie et génétique des populations
Un nombre croissant d’études utilise désormais les codes-barres ADN pour reconstruire l’histoire évolutive d’espèces ou de groupes d’espèces évolutivement proches. Il est ainsi possible, au moyen d’une étude phylogéographique*, d’identifier les mécanismes ayant présidé à la séparation de deux espèces, ou encore de déduire l’histoire de la colonisation d’une région donnée par les populations d’une espèce précise. Ce type d’approche trouve une application particulièrement utile dans l’étude de l’histoire des invasions biologiques à différentes échelles spatiales.
A titre d’exemple, Porco et al. (2013) ont étudié le cas des vers de terre et des collemboles européens qui ont été introduits en Amérique du Nord, où ils sont devenus invasifs. En comparant la diversité génétique observée dans des populations natives et des populations introduites, les auteurs ont montré que la plupart de ces espèces avaient été introduites en Amérique de façon massive et répétée, hormis dans un nombre limité de cas où les populations invasives se sont visiblement constituées à partir d’un nombre plus réduit d’introductions accidentelles.
Dans une autre étude, Valade et al. (2009) ont exploré les relations génétiques entre plusieurs populations européennes du micro-lépidoptère Cameraria ohridella, responsable de la spoliation du feuillage et du dépérissement des marronniers d’Inde dans les parcs et jardins d’Europe de l’Ouest. Ils ont déduit de leur travail que cette espèce invasive est originaire des Balkans, et proposent d’intensifier les études de cette espèce dans cette région de façon notamment à rechercher d’éventuels ennemis naturels qui pourraient être utilisés en lutte biologique*.
Utilisé en routine sur des faunes et dans des régions bien ciblées, le barcoding ADN peut être utilisé comme un système efficace de détection des invasions biologiques. L’avantage de l’approche est qu’elle peut être mise en œuvre par tout un chacun, scientifique ou pas, à n’importe quel endroit du monde et sur n’importe quel groupe d’organismes et n’importe quel type d’échantillon. Elle présente donc un potentiel considérable pour des analyses rapides et automatisées, permettant de déceler l’arrivée d’espèces invasives dans des zones stratégiques, de décrire leur progression en temps réel et de limiter si possible leurs impacts sur leur nouvel environnement.
Analyses de régime alimentaire
La sensibilité élevée des outils moléculaires permet d’envisager leur utilisation pour la caractérisation du régime alimentaire de certains prédateurs à partir de l’ADN contenu dans leur tube digestif ou dans leurs fèces. Bien que basé sur l’utilisation d’un marqueur autre que CO1, le travail de Boyer et al. (2011) illustre parfaitement cette possibilité. Ces auteurs ont ainsi été capables d’identifier certaines proies consommées par un escargot prédateur de vers de terre en comparant les séquences obtenues à partir des déjections de cette espèce à celles d’une librairie de référence. Rougerie et al. (2011) ont également pu identifier les hôtes utilisés par des guêpes parasitoïdes pendant leur stade larvaire à partir d’ADN de l’hôte ayant persisté dans le tube digestif des adultes après la métamorphose (Figure 6). Ces différents résultats ouvrent de nouvelles voies pour la recherche en étendant le champ d’application du barcoding ADN à l’étude des réseaux trophiques et des implications évolutives des interactions proies/prédateurs ou hôtes/parasites.
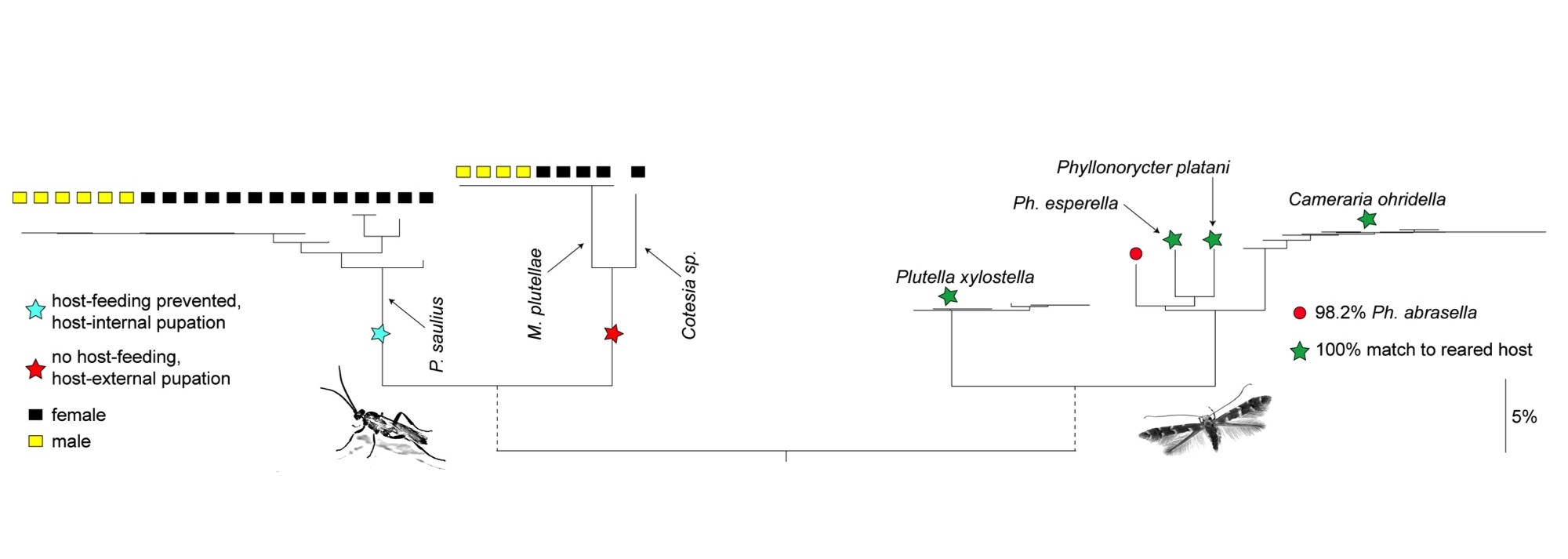
Figure 6. Utilisation des codes-barres ADN pour reconstruire des réseaux trophiques. Cet arbre de distances génétiques est construit à partir de codes-barres ADN obtenues à partir d’ADN extrait des abdomens de guêpes parasitoides. En utilisant des amorces dont l’amplification est sélective, les auteurs ont pu obtenir d’une part les codes-barres des guêpes (partie gauche de l’arbre) et d’autre part ceux des papillons utilisés comme hôtes par les larves de ces guêpes (partie droite). (Figure d’après Rougerie et al., 2010).
Barcoding environnemental
Avec l’avancée des séquenceurs de nouvelle génération, l’utilisation de séquences obtenues à partir de l’ADN total issu d’échantillons environnementaux (par exemple un échantillon de sol ou d’eau, ou encore un ensemble d’invertébrés non triés obtenus dans un piège) est en train de révolutionner les études sur la biodiversité (Hajibabaei 2012; Taberlet et al. 2012). Le barcoding environnemental s’appuie sur la capacité de séquençage parallèle massif des séquenceurs de dernière génération, tels que le « Roche 454 Genome Sequencer » ou « Illumina », qui permettent d’obtenir en une seule analyse des centaines de milliers de séquences à partir de ces échantillons environnementaux.
Même si la longueur de ces séquences est inférieure à celle que l’on peut obtenir à partir de techniques de séquençage classiques, il a été démontré que des fragments d’ADN plus courts (des « mini-barcodes ») restent efficaces pour réaliser des identifications d’espèces. Le potentiel de la méthode est considérable puisque, couplée à l’utilisation de librairies de référence de codes-barres ADN, elle permet d’obtenir l’identité de l’ensemble des espèces présentes dans les échantillons, ceci de façon beaucoup plus rapide que ce qui pourrait être envisagé avec des identifications morphologiques classiques ou même avec du barcoding ADN standard, permettant ainsi d’envisager des études très complète de la biodiversité à l’échelle de l’écosystème (Hajibabaei 2012; Yu et al. 2012).
Estimations de biodiversité
Le barcoding ADN et le barcoding environnemental peuvent également être utilisés pour décrire la diversité et la structure de communautés écologiques. Les séquences obtenues peuvent être utilisées pour définir des « clusters génétiques* », utilisés comme unités taxonomiques moléculaires opérationnelles (MOTU ou OTU). L’utilisation du nombre de ces unités pour estimer la richesse en espèces de communautés d’invertébrés constitue une opportunité intéressante pour étudier des groupes d’organismes ou des régions géographiques particulièrement méconnues.
D’un autre côté, même pour des groupes d’organismes beaucoup mieux connus, l’utilisation de cette approche permet de traiter de façon équivalente l’ensemble des individus d’un échantillon donné et de prendre en compte les cas probables de diversité cryptique, permettant ainsi une plus grande précision dans les résultats obtenus. Young et al. (2012) ont par exemple utilisé cette approche pour décrire les communautés d’acariens du sol dans la station de recherche de Churchill (Manitoba, Canada). Ils ont obtenu près de 6300 séquences représentant environ 900 MOTUs, et ont utilisé leurs résultats pour décrire la composition des communautés (distribution des MOTUs au sein des principaux ordres et familles d’acariens). Ils ont ainsi estimé à près de 1200 le nombre d’espèces d’acariens du sol dans cette localité.
Couplées à des coordonnées géo-référencées, les données collectées dans les campagnes de barcoding ADN permettent également d’aborder la question de la distribution de la diversité des communautés d’invertébrés à des échelles spatiales plus large. L’existence de patrons très généraux (gradients latitudinaux ou altitudinaux, relations entre richesse spécifique et paramètres climatiques, points chauds de biodiversité, etc.) peut ainsi être testée, alors que de telles études ne sont disponibles à l’heure actuelle que pour un nombre réduit de groupes d’invertébrés (Decaëns 2010).
Sciences citoyennes, éducation et recherche
Certains auteurs ont souligné le fort potentiel du barcoding ADN, combiné ou non à des approches basées sur la reconnaissance morphologique des espèces, pour le développement des sciences citoyennes et de programmes éducatifs (Janzen & Hallwachs 2011; Santschi et al. 2013, et voir le regard n°11 sur la science participative). Intégré au sein des offres de formations académiques ou citoyennes, le barcoding ADN se révèle un excellent outil pour articuler les programmes pédagogiques et les enjeux de la recherche actuelle. Au Costa Rica, Daniel H. Janzen et Winnie Hallwachs ont ainsi mis en place un programme participatif impliquant les populations locales de la région de Guanacaste (Janzen & Hallwachs 2011). En combinant la formation de parataxonomistes* et l’utilisation des codes-barres ADN, ils ont permis la réalisation d’un inventaire considérable de la diversité des lépidoptères de cette zone, et ont fourni une quantité extraordinaire de connaissances relatives à la taxonomie et à l’écologie de ce groupe d’organismes (Hebert et al. 2004; Janzen & Hallwachs 2011; Janzen et al. 2013).
Dans un contexte universitaire, les auteurs de ce regard ont récemment participé à la mise en place de l’école de terrain ECOTROP, qui réunit chaque année depuis 2011 des étudiants africains et français dans le parc national de La Lopé au Gabon, avec pour objectif de les former aux pratiques de terrain couramment utilisées en écologie et en paléo–écologie tropicale (coordinateurs : D. Sebag, R. Oslisly et T. Decaëns). Au sein de ce dispositif, le barcoding ADN est utilisé pour décrire la diversité de plusieurs groupes d’invertébrés (faune du sol, insectes) dans les différents écosystèmes de la zone d’étude (Figure 7).

Figure 7. Le barcoding dans les offres de formation scientifique : l’école de terrain ECOTROP propose une formation en écologie tropicale impliquant les étudiants dans un projet de description et de suivi de la biodiversité des invertébrés du Parc National de La Lopé au Gabon. Le barcoding ADN y est utilisé pour s’affranchir du déficit taxonomique de façon à estimer la richesse et à décrire la structure des communautés écologiques dans différents écosystèmes caractéristiques de la zone d’étude.
Conclusion
L’intérêt du barcoding ADN pour l’étude des invertébrés terrestres réside principalement dans sa capacité à lever au moins en partie le déficit taxonomique qui pèse sur ces organismes. Les codes-barres ADN peuvent renforcer les décisions prises pour les actes de nomenclature, tels que les descriptions formelles de nouvelles espèces ou les révisions taxonomiques. Seuls ou associés à des approches environnementales, ils représentent également un outil très prometteur pour la description des patrons spatio-temporels de la biodiversité à des échelles allant du local au global.
.
Glossaire
Caste, chez les insectes et autres invertébrés sociaux : groupe d’individus partageant la même fonction dans la colonie (par exemple les ouvrières ou les soldats).
Cluster génétique : Ensemble d’individus se caractérisant par une similitude génétique suffisante pour qu’ils puissent être considérés comme appartenant à la même espèce ou sous-espèce.
Lutte biologique : Méthode de lutte contre un ravageur ou une plante indésirable dans une culture au moyen d’organismes naturels (par exemple des parasites ou des prédateurs naturels qui vont contrôler l’espèce cible).
Déficit taxonomique (en anglais « Linnean shortfall », « taxonomic impediment ») : Fait référence aux lacunes dans nos connaissances de la biodiversité pour un groupe d’organismes. Peut se calculer comme le rapport entre le nombre d’espèces supposées exister dans ce groupe et le nombre d’espèces actuellement connues.
Diversité cryptique : Diversité non apparente au plan morphologique d’espèces parfaitement individualisées au plan génétique.
Gène mitochondrial : Gène contenu dans le matériel génétique propre aux mitochondries (organites cellulaires dédiés à la respiration), par opposition aux gènes nucléaires caractéristiques du noyau des cellules (eucaryotes).
Génome : Ensemble du matériel génétique d’un individu ou d’une espèce, codé sous forme de séquences d’ADN dans ses chromosomes.
Introgression : Désigne la dispersion des gènes d’une espèce à l’intérieur du pool génétique d’une autre espèce par le biais d’une hybridation entre ces deux espèces.
Parataxonomie : Discipline s’intéressant aux caractères utiles pour l’identification d’organismes par des non-spécialistes. Les « parataxonomistes » sont des personnes n’ayant pas de formation scientifique approfondie en systématique, mais capables de comprendre et d’utiliser des protocoles d’identification d’espèces pour collaborer sur le terrain aux inventaires systématiques et autres recherches sur la biodiversité, éventuellement dans le cadre d’observatoires citoyens et de programmes de sciences participatives (cf. le regard n°11).
PCR : Abréviation du terme anglais « polymerase chain reaction » (amplification en chaîne par polymérisation en français). Il s’agit d’une méthode de biologie moléculaire permettant de dupliquer en grand nombre une séquence d’ADN ou d’ARN connue, à partir d’une faible quantité initiale.
Phylogéographie : Discipline scientifique ayant pour objet l’étude des principes et processus qui gouvernent la distribution des espèces et des subdivisions intra-spécifiques (par exemple les sous-espèces).
Pseudogène : Désigne un gène inactif au sein d’un génome, du fait d’altérations génétiques le rendant non-fonctionnel et donc incapable de conduire à l’expression d’une protéine. Les pseudogènes peuvent être problématiques pour le barcoding ADN lorsque l’on retrouve dans le génome nucléaire des copies inactives, complètes ou partielles, de gènes mitochondriaux (aussi appelées « numts »).
Séquenceur de nouvelle génération : Equipement scientifique permettant le séquençage à haut débit de l’ADN contenu dans un organisme ou dans un échantillon environnemental (échantillon d’eau ou de sol par exemple).
Spécimen type : Spécimen de référence attaché à un nom scientifique, à partir duquel une espèce vivante ou fossile a été décrite. Il existe différentes catégories de spécimens types, soumises à des codes internationaux de nomenclature.
Taxonomie : Science qui a pour objet de décrire les organismes vivants et de les regrouper en entités, appelées taxons, afin de les identifier, les nommer et les classer.
Bibliographie et sites Internet :
Bibliographie
Boyer S., Yeates G.W. et al. (2011). Molecular and morphological analyses of faeces to investigate the diet of earthworm predators: Example of a carnivorous land snail endemic to New Zealand. Pedobiologia, 54, Supplement, S153-S158.
Condon M.A., Scheffer S.J., Lewis M.L. & Swensen S.M. (2008). Hidden neotropical diversity: Greater than the sum of its parts. Science, 320, 928-931.
Decaëns T. (2010). Macroecological patterns in soil communities. Global Ecology And Biogeography, 19, 287-302.
Decaëns T., Porco D., Rougerie R., Brown G.G. & James S.W. (2013). Potential of DNA barcoding for earthworm research in taxonomy and ecology. Applied Soil Ecology, 65, 35- 42.
Gullan P.J. & Cranston P.S. (2010). The Insects: An Outline of Entomology. Wiley-Blackwell.
Hajibabaei M. (2012). The golden age of DNA metasystematics. Trends in Genetics, 28, 535-537.
Hebert P.D.N., Cywinska A., Ball S.L. & deWaard J.R. (2003). Biological identifications through DNA barcodes. Proceedings of the Royal Society of London B, 270, 313-321.
Hebert P.D.N., Penton E.H., Burns J.M., Janzen D.H. & Hallwachs W. (2004). Ten species in one: DNA barcoding reveals cryptic species in the neotropical skipper butterfly Astraptes fulgerator. Proceedings of the National Academy of Sciences of the United States of America, 101, 14812-14817.
James S.W., Porco D. et al. (2010). DNA Barcoding Reveals Cryptic Diversity in Lumbricus terrestris L., 1758 (Clitellata): Resurrection of L. herculeus (Savigny, 1826). Plos One, 5, e15629.
Janzen D.H. (2004). Now is the time. Philosophical Transactions of the Royal Society of London Series B-Biological Sciences, 359, 731-732.
Janzen D.H. (2010). Hope for tropical biodiversity through true bioliteracy. Biotropica, 42, 540‐542.
Janzen D.H. & Hallwachs W. (2011). Joining inventory by parataxonomists with DNA Barcoding of a large complex tropical conserved wildland in Northwestern Costa Rica. PLoS One, 6, e18123.
Janzen D.H., Hallwachs W. et al. (2013). What happens to the traditional taxonomy when a wellknown tropical saturniid moth fauna is DNA barcoded? Invertebrate Systematics, 26, 478–505.
Porco D., Bedos A. et al. (2012a). Challenging species delimitation in Collembola: cryptic diversity among common springtails unveiled by DNA barcoding. Invertebrate Systematics, 26, 470-477.
Porco D., Decaëns T. et al. (2013). DNA Barcoding as a monitoring tool for biological invasions in soil: the case study of European earthworms and collembolans invasion in North America. Biological Invasions, 15, 899-910.
Porco D., Potapov M. et al. (2012b). Cryptic diversity in the ubiquist species Parisotoma notabilis (Collembola, Isotomidae): A long-used chimeric species? Plos One, 7, e46056.
Primack R.B. (2000). Primer of Conservation Biology. Sinauer Associates, Sunderland.
Puillandre N., Bouchet P. et al. (2012). New taxonomy and old collections: Integrating DNA barcoding into the collection curation process. Molecular Ecology Resources, 12, 396-402.
Ratnasingham S. & Hebert P.D.N. (2007). The Barcode of Life Data System (www.barcodinglife.org). Molecular Ecology Notes, 7, 355–364.
Rougerie R., Naumann S. & Nässig W.A. (2012). Morphology and molecules reveal unexpected cryptic diversity in the enigmatic genus Sinobirma Bryk, 1944 (Lepidoptera: Saturniidae). PLOSone, 7, e43920.
Rougerie R., Smith M.A. et al. (2011). Molecular analysis of parasitoid linkages (MAPL): Gut contents of adult parasitoid wasps reveal larval host. Molecular Ecology, 20, 179-186.
Santschi L., Hanner R.H. et al. (2013). Barcoding life’s matrix: Translating biodiversity genomics into high school settings to enhance life science education. PLoS Biol, 11, e1001471.
Stoeckle M.Y. & Hebert P.D.N. (2008). Barcode of life. Scientific American, Oct 2008, 82-88.
Taberlet P., Coissac E., Hajibabaei M. & Rieseberg L.H. (2012). Environmental DNA. Molecular Ecology, 21, 1789-1793.
Valade R., Kenis M. et al. (2009). Mitochondrial and microsatellite DNA markers reveal a Balkan origin for the highly invasive horse-chestnut leaf miner Cameraria ohridella (Lepidoptera, Gracillariidae). Molecular Ecology, 18, 3458-3470.
Wilson E.O. (2002). The Future of Life. New York.
Young M.R., Behan-Pelletier V.M. & Hebert P.D.N. (2012). Revealing the hyperdiverse mite fauna of subarctic Canada through DNA barcoding. Plos One, 7, e48755.
Yu D.W., Ji Y. et al. (2012). Biodiversity Soup: metabarcoding of arthropods for rapid biodiversity assessment and biomonitoring. Methods in Ecology and Evolution, 3, 613-623.
Et ces “regards” en ligne, sur des sujets connexes :
Barot S. et F. Dubs, 2012. Les écosystèmes du sol. Regards et débats sur la biodiversité, SFE, regard n°28, 17 février 2012.
Corbara B., 2011. En quête d’espèces : A quoi servent les expéditions scientifiques ? Regards et débats sur la biodiversité, SFE, regard n°23, 3 novembre 2011.
Doré T., 2011. La biodiversité, atout pour l’agriculture. Regards et débats sur la biodiversité, SFE, regard n°24, 22 novembre 2011.
Teyssèdre A., 2010. Les services écosystémiques, notion clé pour explorer et préserver le fonctionnement des (socio)écosystèmes. Regards et débats sur la biodiversité, SFE, regard n°4, 25 octobre 2010.
Teyssèdre A. et D. Couvet, 2011. Biodiversité et science participative. Regards et débats sur la biodiversité, SFE, regard n°11, 6 février 2011.
Sites Internet
– Site du « Barcode of Life Data Systems » (BOLD) : www.boldsystems.org/
– Site du consortium « Barcode of Life » (CBOL) : http://www.barcodeoflife.org/
– Site du « European Consortium for the Barcode of Life »: http://www.ecbol.org/
– Site du projet « international Barcode of Life » (iBOL): www.iBOL.org/
– Site du Canadian Centre for DNA barcoding donnant accès aux protocoles : http://www.ccdb.ca/
– Sites de campagnes de barcoding de différents groupes d’organismes : http://www.formicidaebol.org/, http://www.trichopterabol.org/, http://earthwormbol.org/index.php, http://www.lepbarcoding.org/
– Blogs et plateformes sociales dédiés au barcoding: http://phe.rockefeller.edu/barcode/blog/,
http://dna-barcoding.blogspot.fr/, http://connect.barcodeoflife.net/
– Site de l’école de terrain ECOTROP : http://www.ecotrop.com/
—-
Article édité par Anne Teyssèdre
Le séquençage haut-débit génère maintenant des séquences ADN amplifiées (« amplicons ») assez longues pour pouvoir reconstruire des arbres phylogénétiques (phylogénies).
Deux références récentes:
http://onlinelibrary.wiley.com/doi/10.1002/ece3.508/full
http://onlinelibrary.wiley.com/doi/10.1111/1755-0998.12173/abstract
Bonjour,
et merci pour cet intéressant « regard » sur le barcoding ADN. J’ai une question sur les applications de cette méthode à la détermination d’espèces jumelles/cryptiques : Qu’est-ce qui permet d’affirmer que le gène CO1, utilisé notamment pour distinguer les espèces jumelles (non différentiables au plan morphologique), est un critère d’espèce ?
En d’autres termes, lorsque deux « formes » jumelles d’invertébrés diffèrent par leur gène CO1, sur quelle base affirmer qu’il s’agit de deux espèces vraies, et non pas seulement de deux variants génétiques, ou deux sous-espèces ?
[Rappel: le gène CO1 est impliqué dans le métabolisme aérobie, donc a priori sans grand rapport avec l’isolement reproducteur ou la différentiation entre espèces.]
Il me semble que deux formes jumelles différant par leur gène CO1 doivent d’abord être reconnues comme deux espèces vraies, sur un ou plusieurs autres critères indépendants, avant d’utiliser leur différence génétique sur ce gène mitochondrial comme critère de différentiation spécifique (= entre espèces). Est-ce bien le cas ?
Bien cordialement,
Anne
Bonjour Anne,
Désolé de répondre à ton message avec un peu de retard, nous revenons justement de la récente 5ème conférence internationale de barcoding à Kunming (Chine). J’en profite pour mettre un lien vers le site de la conférence pour que ceux qui seraient intéressés puisse aller jeter un oeil au programme qui était fort intéressant avec un virage net vers les applications de l’outil en écologie (cf. session plénière du mercredi notamment) et l’intégration des méthodes NGS : http://www.dnabarcodes2013.org
Pour en revenir à ta question, tu as raison, on ne peut affirmer que 2 échantillons appartiennent à 2 espèces distinctes sur la seule base de séquences distinctes du gène COI. En effet ce gène est variable à l’intérieur des espèces, et il n’y a pas de seuil de divergence précis permettant d’affirmer que l’on a affaire à des espèces distinctes et non à de la variation intraspécifique.
En principe, les taxonomistes qui utilisent les codes-barres ADN vont aborder les descriptions d’espèces nouvelles de manière intégrative en cherchant à corroborer les résultats du barcoding par des caractères morphologiques, des caractéristiques écologiques (plantes hôtes par exemple) ou des marqueurs moléculaires indépendants (génome nucléaire). Bien souvent, c’est aussi l’inverse qui arrive, et les divergences génétiques observées pour COI viennent confirmer des différences morphologiques subtiles observées auparavant.
Ceci étant dit, tu as peut-être vu que les méthodes de délimitation automatique des espèces sont en train de se multiplier dans la littérature. Elles ont l’avantage de pouvoir être appliquées pour des groupes et dans des situations où un recours à la corroboration morphologique n’est pas envisageable (ex. groupe souffrant d’un fort déficit taxonomique, ou échantillons avec beaucoup d’individus traités par NGS). Les approches multigéniques sont alors à recommander, mais il n’est pas du tout évident de trouver un gène nucléaire dont la variabilité est satisfaisante. Aussi, plusieurs méthodes ont été développées pour un seul marqueur et notamment le code-barres ADN (ex. ABGD, GMYC, RESL/BINs). Je ne vais pas rentrer ici dans les détails car ce post est déjà suffisamment long, mais ces méthodes marchent relativement bien et peuvent être testées empiriquement sur des groupes où la taxonomie traditionnelle peut corroborer les résultats. Ensuite on peut franchir le pas et les appliquer sur des faunes totalement inconnues, non décrites, etc. (cf. par exemple ce papier que nous avons récemment publié dans MER : http://onlinelibrary.wiley.com/doi/10.1111/1755-0998.12178/abstract)
Cordialement,
Rodolphe
Modifier
Bonjour,
En plus de son intérêt d’un point de vue qualitatif, est-ce que cette méthode permet des approches quantitatives (estimation du nombre d’individus présents dans un échantillon) ? En d’autres termes, est-ce que cette méthode peut remplacer les heures passées sur la loupe binoculaire lors du tri d’invertébrés (pièges Barber par exemple) ?
Merci d’avance.
Sylvain G.
Salut Sylvain,
Oui en effet, ce type d’outil est voué à réduire le temps de travail sous la loupe. Pour cela deux solutions:
– la force brute, soit séquencer en Sanger (séquençage classique d’un spécimen à la fois) tous les individus de l’échantillon,
– les nouvelles techniques de séquençage massif parallèle (Illumina, 454…etc) capables de séquencer toutes les molécules d’ADN dans un mélange, on parle alors de ‘soupe de biodiversité’.
Dans les deux cas il faudra bien sûr comparer chacune des séquences obtenues à une bibliothèque de référence constituée en amont via le séquençage de spécimens dûment identifiés à l’espèce. En l’absence de telles bibliothèques, on pourra obtenir une approximation du nombre d’espèces en appliquant une valeur seuil relative au niveau spécifique pour le groupe étudié.
De plus en plus d’études font appel à cette deuxième méthode plus rapide (car elle ne nécessite pas de manipulation de chacun des spécimens) et moins couteuse. Bien sûr on en est encore aux prémices donc les résultats ne sont pas parfaits en quantitatif, on peut en effet avoir certains biais liés à des niveaux d’affinités variables des oligonucléotides employés par rapport aux différentes espèces ou bien avoir un nombre de mitochondries variable d’une espèce à l’autre. Ceci dit les premiers résultats sont très encourageants, et la méthodologie évolue très vite :
http://onlinelibrary.wiley.com/doi/10.1111/j.2041-210X.2012.00198.x/abstract
http://onlinelibrary.wiley.com/doi/10.1111/j.1755-0998.2009.02611.x/pdf
http://www.gigasciencejournal.com/content/2/1/4
Cordialement
David
Bonjour,
Très intéressant regard sur l’outil de barcoding, bravo ! En tant qu’amateur en entomologie et ayant une spécialité sur les Coléoptères Erotylidae néotropicaux, j’ai hâte de pouvoir accéder à cet outil pour l’aide en systématique. C’est a ma portée financièrement, soit personnellement soit en montant un projet (si j’estime bien environ 5 à 10 euros par individu – soit maximum 1000 euros pour une plaque de 95 échantillons). Mais le problème serait plutôt de trouver à m’inscrire dans un programme ou de trouver un labo !
Et nous n’en sommes qu’au début de ces applications. L’écologie de beaucoup d’insectes européens est encore mal ou pas du tout connue, alors imaginez pour des consommateurs de champignons de la forêt amazonienne! Justement, avec les nouveaux outils qui arrivent (cf l’article Ultra-deep sequencing enables high-fidelity recovery of biodiversity for bulk arthropod samples without PCR amplification, par exemple), on pourra faire d’une pierre deux coups en analysant les contenus abdominaux et avoir le barcode de l’insecte et de ce qu’il a ingéré. Bon, je ne sais pas si ca marche pour les champignons et/ou si on peut extraire les deux types de barcodes en même temps, mais il suffit probablement d’être patient.
En tant qu’archéologue, je suis fort curieux de voir ce qu’on pourra extraire en faisant du metabarcoding de sédiments avec une bonne conservation (sous l’eau et organiques). Dans ce genre de milieu de conservation, feuilles, graines, bois et insectes sont conservés, ce qui potentiellement pourrait permettre de reconstruire des environnements anciens. Cela a déjà été fait pour des milieux gelés récents et donne de très bons résultats. Les travaux de l’équipe Taberlet sur les prairies alpines tendent à montrer que même avec des sols récents on doit pouvoir remonter un peu dans le temps.
Je m’étonne qu’un archéologue (français) soit tout à la fois entomologiste amateur spécialisé dans l’étude de certains Coléoptères néotropicaux, intéressé par la taxonomie, l’écologie et l’archéozoologie des arthropodes, et prêt à financer lui-même ses recherches dans ces domaines… Si c’est bien le cas, bravo!
Bonjour,
Juste une rapide réaction au post en effet très éclectique de Jean-Hervé, qui voit juste sur ses différents points:
– Le barcoding est bien un outil complémentaire pour le taxonomiste, « amateur » ou pro. Au sein de la communauté des lépidoptéristes (mais ce n’est bien sûr pas la seule) l’outil est déjà très largement intégré aux pratiques taxonomiques actuelles. De nombreux taxonomistes ont pu profiter pendant quelques années de financements iBOL qui leur ont permis à la fois de contribuer à la construction des librairies de référence (80,000 espèces déjà barcodées !) et de bénéficier de l’apport de cet outil pour la délimitation des espèces.
Depuis 2010, iBOL ne peut plus couvrir la totalité des coûts du barcoding d’échantillons, mais tout taxonomiste souhaitant intégrer les barcodes à sa boîte à outil peut toujours le faire dans le cadre de ce projet et bénéficier des infrastructures (labos, bases de données et outils d’analyse) en place. A titre informatif, et à moins d’avoir accès à un autre labo, le coût actuel pour du barcoding « standard » en passant par le CCDB (Canadian Centre for DNA BArcoding) est de 1000CAD (715 euros) par plaque de 95 échantillons. C’est un coût qui peut être intégré dans un projet de recherche, mais il n’est pas si rare de voir des taxonomistes « amateurs » le prendre maintenant en charge par eux-même pour bénéficier de l’outil.
… Si Jean-Hervé veut se lancer dans un projet sur les Erotylidae néotropicaux, je peux l’aiguiller hors-liste pour se lancer 🙂
– Le second point souligne le potentiel de ces approches en écologie, largement évoqué dans ce « Regard ». La documentation des réseaux trophiques avec ce type d’outil est très prometteuse et le type d’application suggérée pour le régime alimentaire des Erotylidae est déjà à portée de main et utilisée dans d’autres groupes.
– Finalement, des approches de type « métabarcoding » sont en effet déjà utilisées sur des sédiments ou carottes de glace pour des études de paléoécologie et offrent une vision unique de la biodiversité du passé, réservant sans doute de belles surprises dans les années à venir, avec probablement d’intéressantes découvertes sur les dynamiques temporelles et spatiales des communautés et des populations, et des inférences importantes sur les effets des changements globaux et surtout de l’évolution du climat.
Salutations,
Rodolphe
Bonjour et merci,
Ces multiples casquettes sont bien miennes, même si elles sonnent un peu comme un inventaire à la Prévert et évoquent un joyeux touche à tout. J’assume cet aspect et cette dispersion apparente qui dérivent d’un intérêt pour la nature, l’homme, l’homme dans la nature et leurs histoires communes…
Cela s’est traduit dans mon métier ( je suis archéozoologue professionnel à l’INRAP) et dans mes loisirs (entomologiste amateur, président de l’association régionale et membre du CSRPN – comité supérieur régional de protection de la nature – dans ma région en Picardie). En tant qu’entomologistes amateurs nous consacrons beaucoup de temps et d’argent à notre passion (matériel, déplacement…). Alors une fois que l’on a intégré le fait que barcoding métabarcoding sont des outils complémentaires et vont ouvrir en grand de nouvelles opportunités, il me paraît logique de mettre/trouver un peu d’argent pour des objectifs personnels et collectifs. La troisième génération de séquenceur pointe seulement le bout de son nez et les prix vont continuer à s’effondrer (pas besoin de se nommer nostradamus pour le prédire). L’accès à cette technique va devenir de plus en plus aisé. Ces techniques vont faire exploser notre compréhension de la biodiversité, de l’écologie et beaucoup de gens sont en train d’en prendre conscience. Un amateur éclairé ne peut donc qu’être tenté de participer à cette dynamique et mettre à profit la formidable opportunité qui apparaît.
Coté professionnel, nous y sommes déjà en partie quand on voit que de l’extraction d’ADN des os d’animaux issus de sites archéologiques aboutit à des articles sur la couleur du pelage des anciens animaux domestiques et son évolution ou sur l’histoire de la domestication et des migrations… Mais là encore ce n’est que le début et je pense que le métabarcoding quand on arrivera à l’appliquer à l’archéologie va révolutionner nos approches.